
近日,国际综合性学术期刊《Nature Communications》发表我室周昆研究员及其合作者的研究成果“V- and VL-scores unveil viral signatures and origins of protein families”。该研究聚焦病毒基因组与蛋白质的研究工作,提出了两种无需注释的定量指标——V-score 与 VL-score,用于衡量蛋白家族及基因组的“病毒相似度”,并构建了开放访问的搜索数据库“V-Score-Search”。该研究为解析病毒特征及其与宿主相互作用提供了新视角,在基因组学、生物技术及微生物学领域具有广泛的应用前景和重要的科学意义。
病毒是生物圈中不可或缺的组成部分。凭借在微生物组和生态系统中的极高丰度与高度遗传多样性,病毒能够调控种群数量、促进营养循环、增加遗传多样性,并驱动病毒-宿主共进化动态。尽管病毒至关重要,但实验室培养困难,导致大量病毒仍未被认知。计算方法作为重要的补充工具,被广泛用于研究病毒基因组和蛋白质。解析病毒基因组与蛋白质,对于理解其多样性及其在生态系统中的功能至关重要,也将推动疫苗、噬菌体疗法等生物技术的发展。
传统上,病毒特异性基因(如衣壳蛋白等标志基因)被视为病毒基因组的特征,用于病毒基因组的鉴定与分类。然而,标志基因仅占病毒基因组的一小部分。基因组或宏基因组片段通常不含标志基因,使得传统的以基因为中心的方法难以识别和分类病毒。因此,大量病毒基因组未被识别,造成信息严重缺失,亟需突破当前在病毒发现与蛋白质注释方面的局限。
注释病毒基因并预测其功能,可为理解病毒序列和蛋白家族的特性提供线索。该研究指出,结合功能注释分析整个病毒基因组,有望突破常规识别出创新的病毒特征信号,即便在片段化状态下也依然有效。为此,该研究引入V-score和VL-score的概念——用于区分病毒与非病毒蛋白家族及基因组的量化指标,作为“病毒特征”(图1)。该研究系统性地进行了计算和测试,并公开发布了覆盖五个主要蛋白质数据库(KEGG、Pfam、eggNOG、PHROG和VOG)的V-score及对数尺度VL-score。通过构建开放搜索数据库(https://anantharamanlab.github.io/V-Score-Search/),该研究使这些指标得到了广泛应用。综合分析表明,这些评分为多个病毒研究难题提供了强大的解决方案,包括精确的病毒序列识别、改进的原噬菌体检测以及准确的辅助代谢基因注释。
以下结果将展示V-score和VL-score如何作为一种通用的病毒蛋白识别指标,规避传统标志基因方法的局限,显著超越现有方法。
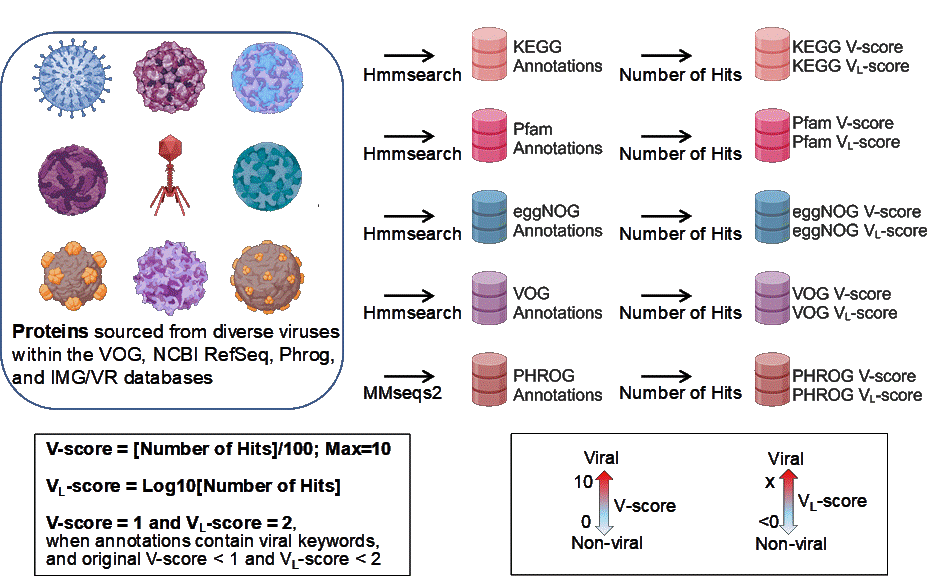
图 1. V-score 与 VL-score 的概念和生成流程图。图中展示了该研究所用多种病毒中具有代表性的九个病毒类群。V-score 和 VL-score 的标尺分别以从 0 到 10 和从 <0 到 X 的双向箭头表示,分数低代表非病毒来源,分数高表示病毒来源。
(一)在病毒基因组序列鉴定方面,通过应用基于KEGG、Pfam、VOG或PHROG推导出的、针对不同长度基因组片段的AV-score(Average V-score)阈值,该研究对39个宏基因组共鉴定出13167条低、中、高质量的病毒序列(Contig长度 ≥ 1 kb)(图2a)。其中,2045条序列与依赖病毒特异性标志基因的识别工具geNomad所鉴定的序列重叠。尽管与geNomad(鉴定出17369条)相比,该方法鉴定出的低质量病毒序列较少(10827条),但在中、高质量序列的鉴定方面,该方法显著优于geNomad。基于AV-score的方法鉴定出超过1000条高质量病毒序列,约为geNomad预测数量的七倍。
该研究进一步测试了该方法在原噬菌体鉴定与评估中的潜力,结果显示,PHASTER原噬菌体数据库中的65668个原噬菌体序列中,超过95%的序列被该方法预测为病毒基因组(图2b)。以大肠杆菌及其原噬菌体为例,该研究清晰展示了大肠杆菌原噬菌体与其相邻大肠杆菌宿主序列之间的边界(图2c)。
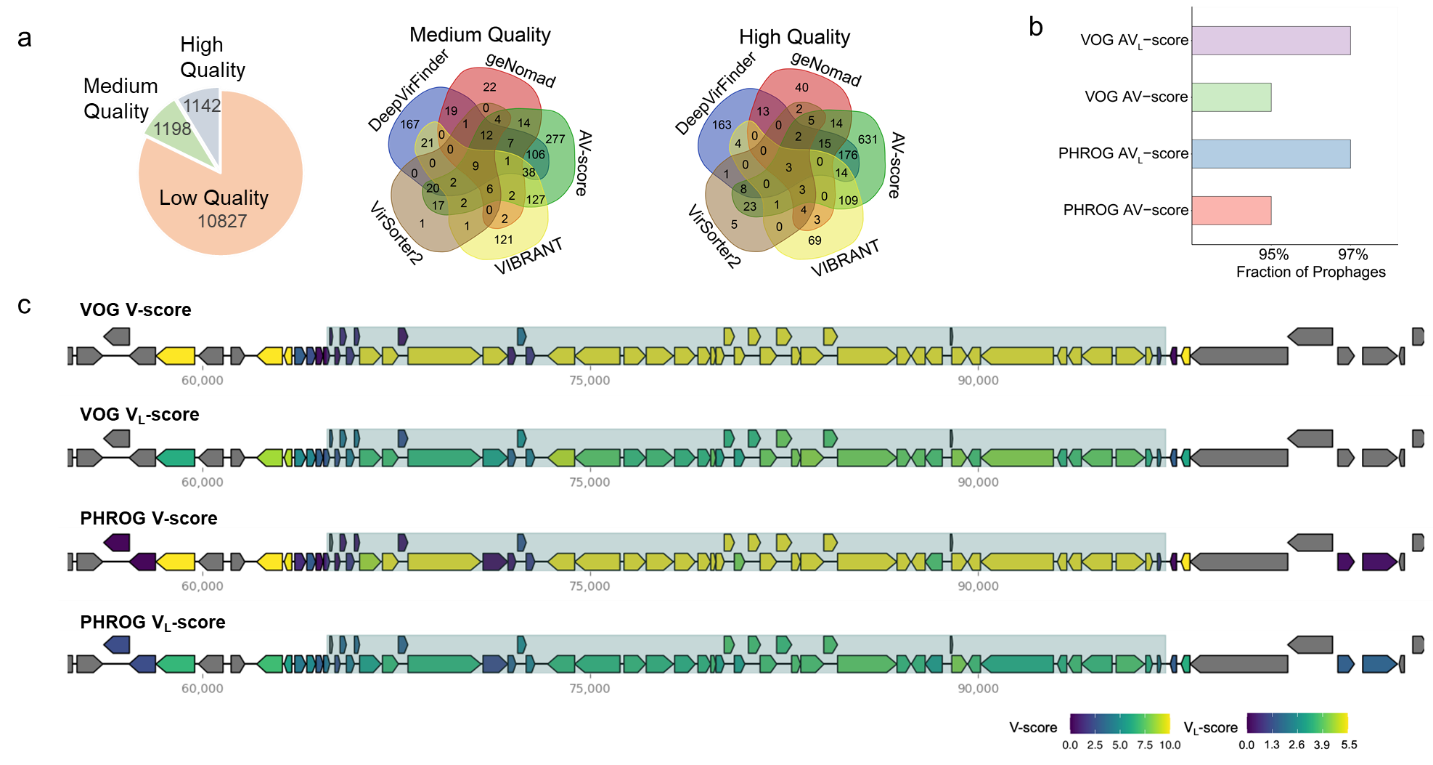
图 2. 应用 V-score、VL-score、AV-score(Average V-score) 和 AVL-score (Average VL-score)进行基因组及宏基因组中的病毒识别。a,使用 AV-score 和 AVL-score 以及四种常用软件识别出的序列数量。针对 CheckV 评估为中高质量且长度 ≥5 kb 的序列,使用维恩图展示了五种方法之间的重叠情况,显示了不同方法鉴定出的共有序列和特有序列的数量。b,数据库中具有 AV-score 和 AVL-score 超过相应判定为病毒样阈值的原噬菌体所占比例。c,已验证的大肠杆菌原噬菌体及其相邻宿主序列中基因的 V-score 和 VL-score 分布。原噬菌体区域以阴影突出显示。
(二)在病毒基因鉴定方面,该研究从5116个病毒基因组中共鉴定出27442个可能具有辅助功能的病毒基因。其中,非代谢类辅助病毒基因占绝大多数(89%),而辅助代谢基因仅占一小部分(11%)(图3a)。鉴定出的辅助病毒基因包括编码多种代谢酶、抗生素抗性蛋白、转运蛋白、DNA/RNA复制蛋白、转座酶/重组酶、核酸酶/内切酶以及未表征/假设蛋白的基因。这些辅助病毒基因发挥多种功能,包括代谢、遗传信息处理、环境信息处理以及细胞过程(图3b)。
与其他现有方法(包括VIBRANT和DRAM-v)相比,该研究在辅助代谢基因(AMG)鉴定方面展现出显著优势:当应用于相同的病毒基因组集时,该研究鉴定出3859个AMG(图3c),而VIBRANT和DRAM-v分别仅鉴定出1261个和1993个AMG(图3c)。值得注意的是,三种方法共同鉴定出的AMG的Pfam结构域或KEGG直系同源项仅占一小部分(图3d),大多数AMG为每种方法所独有。这表明该研究能够揭示现有AMG检测工具常常忽略的新功能,与现有工具形成互补。
该研究发现的一些独特代谢酶包括丝氨酸β-内酰胺酶样超家族、ATP-grasp超家族、N-乙酰转移酶样超家族以及胆碱结合重复超家族(图3e)。此外,该研究在所有KEGG类别中鉴定出的AMG数量均高于VIBRANT(图3d)。这些发现表明基于V-score的方法能够以高精度检测出更多潜在的病毒辅助基因。
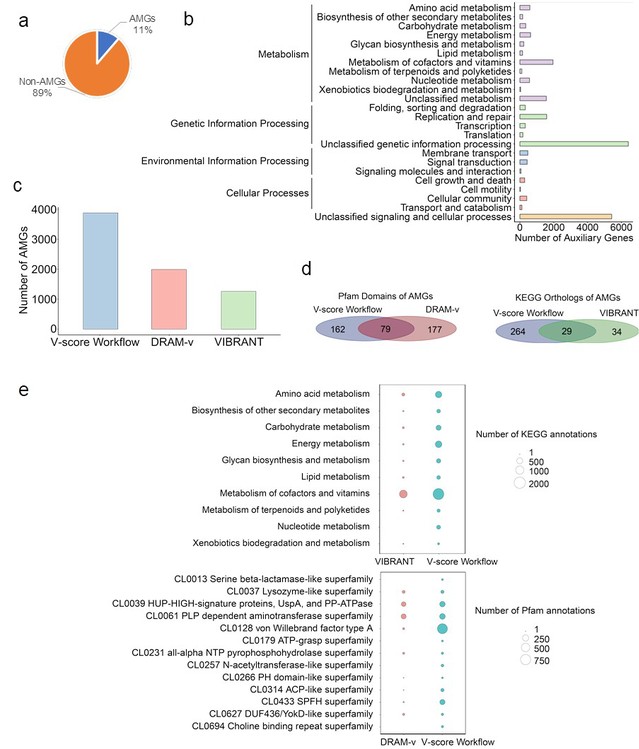
图 3. 该研究中鉴定到的辅助基因及与现有方法的比较。a,辅助基因的组成。AMG:辅助代谢基因。b,通过 V-score 工作流程检测到的、具有注释的辅助基因的潜在功能。c,V-score 工作流程鉴定到的 AMG 数量与DRAM-v 和 VIBRANT方法的对比。d,V-score 工作流程、DRAM-v 和 VIBRANT 鉴定到的 AMG 中共享及特有的 Pfam 结构域或 KEGG 直系同源项韦恩图。e,V-score 工作流程、DRAM-v 和 VIBRANT 鉴定到的 AMG 所具有的 KEGG 或 Pfam 注释数量的比较。需要注意的是,VIBRANT 仅输出包含 KEGG 注释的结果,而 DRAM-v 主要为该研究中鉴定到的 AMG 生成 Pfam 注释。
论文第一作者和共同通讯作者为我室周昆研究员,合作者包括威斯康星大学麦迪逊分校Karthik Anantharaman教授(共同通讯作者),以及威斯康星大学麦迪逊分校研究生James C. Kosmopoulos、Etan Dieppa Colón和Peter John Badciong。该研究受同济大学海洋地质全国重点实验室重点项目、国家自然科学基金以及美国国家科学基金共同资助。
全文链接:https://www.nature.com/articles/s41467-026-72028-0